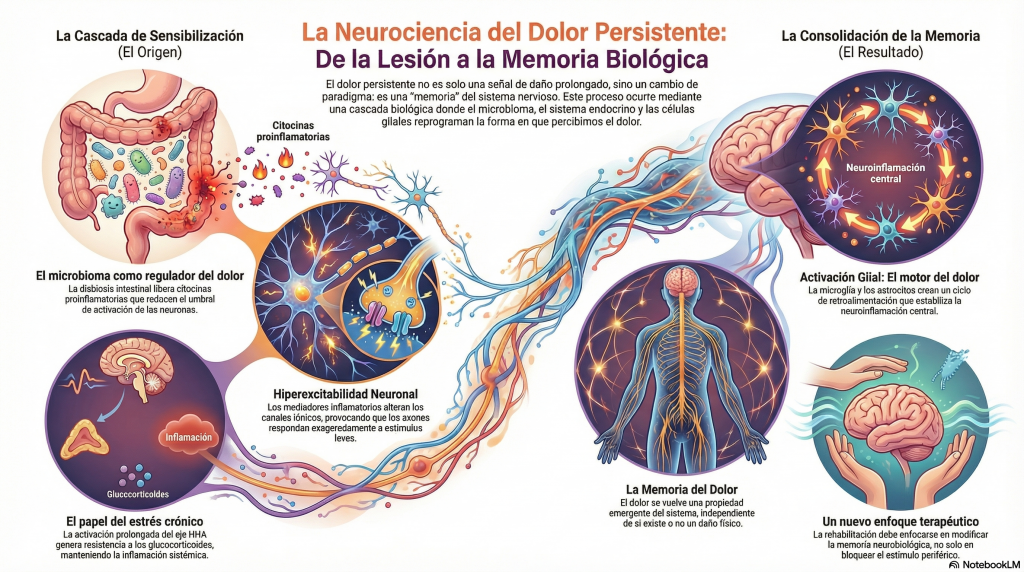
Durante años, el dolor persistente se ha explicado como una prolongación fallida del dolor agudo o como una respuesta exagerada a una lesión periférica. Sin embargo, la neurociencia contemporánea ha desplazado esta visión hacia un modelo en el que el dolor se comporta como un estado funcional aprendido del sistema nervioso. Esta primera entrada, aborda el núcleo de ese cambio de paradigma: cómo la interacción entre microbioma, ejes neuroendocrinos y glía crea las condiciones biológicas necesarias para que el dolor se consolide como memoria.
El dolor persistente, desde la neurociencia contemporánea, no debería poder entenderse como una simple prolongación temporal del dolor agudo o como una señal proporcional de daño tisular mantenido. En la actualidad se conceptualiza como un estado funcional aprendido del sistema nervioso, resultado de cambios duraderos en la excitabilidad neuronal, en la transmisión sináptica y en la interacción entre el sistema nervioso, el sistema inmune y el sistema endocrino.
Este cambio de paradigma implica que el dolor deja de depender exclusivamente del estímulo periférico inicial y pasa a estar determinado por mecanismos centrales de plasticidad neuronal y neuroinflamación capaces de sostener la experiencia dolorosa incluso cuando el daño tisular ha desaparecido o es insuficiente para explicar la magnitud del síntoma.
En este marco adquiere relevancia el microbioma humano, un término que requiere una definición precisa para evitar simplificaciones.
El microbioma no se limita al conjunto de microorganismos que habitan el organismo —lo que se denomina microbiota—, sino que incluye el conjunto de genomas microbianos, los productos metabólicos que estos microorganismos generan y las funciones biológicas que expresan en interacción con el huésped.
Desde una perspectiva funcional, el microbioma puede entenderse como un sistema metabólico y señalizador distribuido que participa activamente en la regulación inmunitaria, endocrina y nerviosa.
El microbioma intestinal constituye el principal nodo de influencia sistémica por varias razones: su enorme densidad microbiana, su elevada diversidad metabólica y su localización estratégica en contacto con el sistema nervioso entérico y con una extensa red inmunitaria.
A través de la fermentación de sustratos dietéticos y del metabolismo de aminoácidos y ácidos biliares, el microbioma produce múltiples metabolitos bioactivos, entre ellos ácidos grasos de cadena corta como el butirato, el propionato y el acetato, así como derivados del triptófano.
Estas sustancias no son subproductos inertes, sino moléculas señalizadoras con capacidad para modular la función inmunitaria, la integridad de barreras biológicas y la actividad del sistema nervioso.
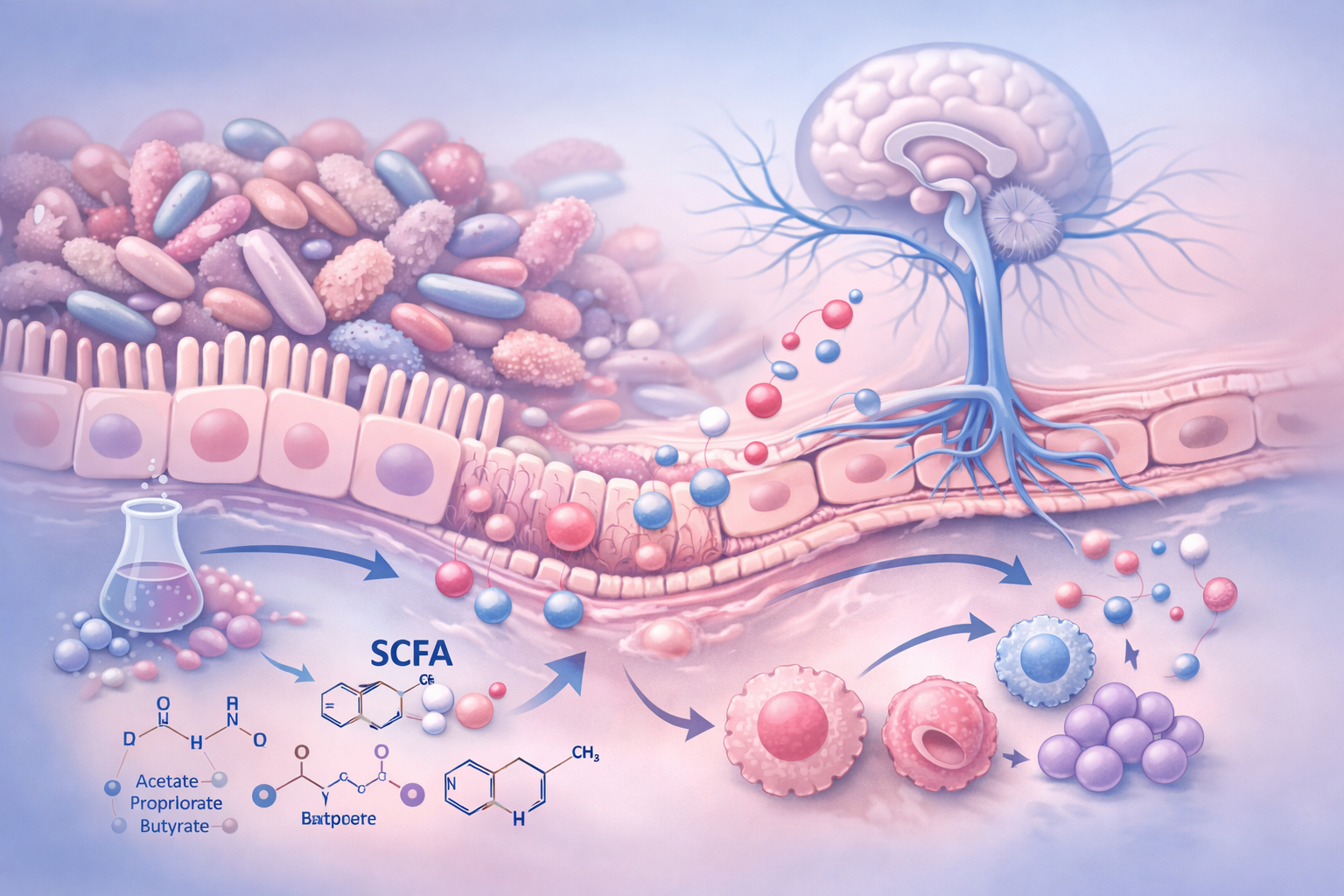
Cuando el equilibrio funcional del microbioma se altera —situación denominada disbiosis— se produce una modificación del tono inflamatorio sistémico. La disbiosis se asocia a un aumento de la permeabilidad intestinal, lo que facilita el paso de componentes microbianos y mediadores inflamatorios a la circulación sistémica.
Este fenómeno incrementa la activación del sistema inmune innato y favorece la liberación de citocinas proinflamatorias como interleucina-1β, interleucina-6 y factor de necrosis tumoral alfa. Estas citocinas no generan dolor por sí mismas, pero modulan la excitabilidad del sistema nervioso, reduciendo el umbral de activación de neuronas nociceptivas tanto periféricas como centrales.
La excitabilidad neuronal hace referencia a la facilidad con la que una neurona genera potenciales de acción.
En el contexto del dolor, esta excitabilidad depende de la función de los canales iónicos presentes en la membrana neuronal, particularmente canales de sodio, potasio y calcio. Las citocinas inflamatorias y otros mediadores derivados de la disbiosis pueden modificar la expresión y la cinética de estos canales, haciendo que los axones nociceptivos respondan de forma exagerada a estímulos de baja intensidad o incluso generen actividad espontánea. Este aumento de excitabilidad constituye uno de los primeros pasos hacia la sensibilización periférica y central.
Los axones, prolongaciones especializadas de las neuronas encargadas de conducir el impulso nervioso, desempeñan un papel clave en este proceso.
En condiciones fisiológicas, la conducción axonal está finamente regulada por la distribución de canales iónicos y por la mielina que recubre muchos axones.
En estados inflamatorios persistentes, esta regulación se altera, facilitando descargas ectópicas y transmisión aberrante de señales nociceptivas. Estas señales, al alcanzar la médula espinal, encuentran un entorno predispuesto a amplificar su impacto debido a cambios previos en la excitabilidad central.
El microbioma también influye de manera decisiva sobre los ejes neuroendocrinos, en particular el eje hipotálamo-hipófisis-adrenal (HHA), principal sistema regulador de la respuesta al estrés. Este eje funciona mediante la liberación secuencial de hormona liberadora de corticotropina desde el hipotálamo, hormona adrenocorticotropa desde la hipófisis y glucocorticoides desde la corteza suprarrenal. Los glucocorticoides ejercen efectos amplios sobre el metabolismo, el sistema inmune y el sistema nervioso. La colonización microbiana temprana programa la reactividad futura de este eje, de modo que alteraciones en dicha colonización se asocian a respuestas exageradas o disfuncionales ante el estrés.
En el dolor persistente, la activación crónica del eje HHA conduce a una exposición prolongada a glucocorticoides que pierde su carácter adaptativo. Aunque en fases agudas estos tienen efectos antiinflamatorios, su presencia sostenida puede inducir resistencia a glucocorticoides, un estado en el que los tejidos dejan de responder adecuadamente a estas hormonas. Esta resistencia favorece la persistencia de la inflamación y contribuye a mantener un entorno neurobiológico propicio para la sensibilización central.
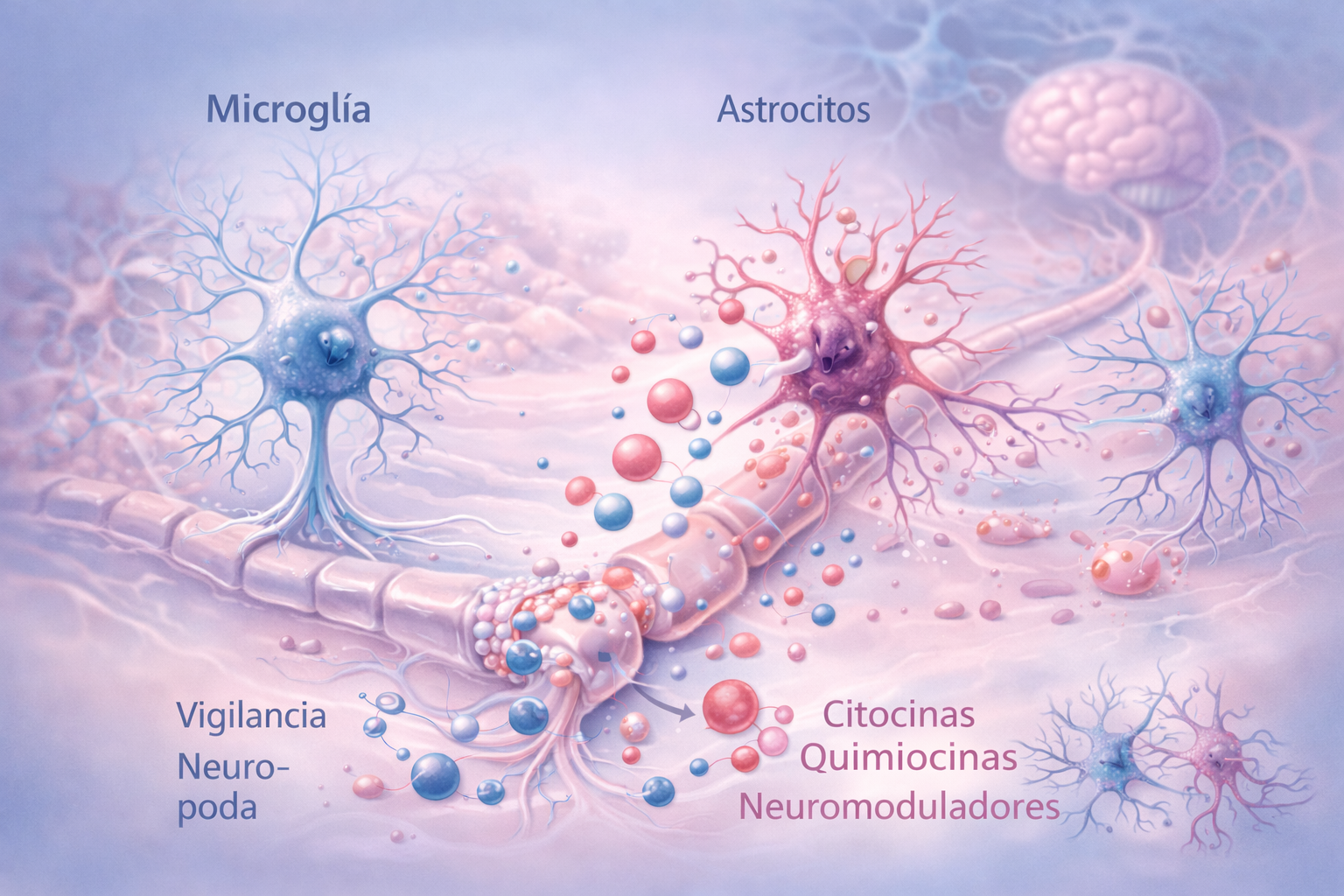
En el sistema nervioso central, estos procesos convergen en la glía, particularmente en la microglía y los astrocitos.
La microglía actúa como sistema inmunitario residente del cerebro y la médula espinal. En condiciones normales participa en la vigilancia del entorno neuronal y en la poda sináptica durante el desarrollo y el aprendizaje, pero si existe inflamación persistente y señalización aberrante, la microglía adopta un estado reactivo caracterizado por la liberación sostenida de citocinas, quimiocinas y neuromoduladores que incrementan la excitabilidad neuronal.
Los astrocitos, por su parte, regulan el microambiente sináptico controlando la recaptación de neurotransmisores excitatorios como el glutamato y aportando soporte metabólico a las neuronas. Cuando se activan de forma crónica, alteran estas funciones y favorecen un entorno excitatorio que prolonga la hiperexcitabilidad neuronal.
La interacción entre microglía y astrocitos establece un circuito de retroalimentación que estabiliza la neuroinflamación y refuerza la plasticidad maladaptativa de los circuitos nociceptivos.
El resultado final de esta cascada es la memoria del dolor, entendida como una huella neurobiológica y maléfica aparentemente sin control y distribuida que se mantiene por cambios duraderos en la excitabilidad neuronal, en la transmisión sináptica y en la activación glial.
El dolor deja de ser una respuesta proporcional al daño y pasa a ser una propiedad emergente del sistema nervioso. En este punto, cualquier intervención eficaz debe dirigirse no solo a la señal periférica, sino a los mecanismos que sostienen esta memoria, lo que abre la puerta a enfoques que integren neurociencia, inmunología, endocrinología y rehabilitación.
Si el dolor persistente se consolida como un estado neurobiológico mantenido por plasticidad y neuroinflamación, la pregunta clínica ya no puede ser únicamente “qué bloquea el dolor”, sino “qué intervenciones son capaces de modificar esa memoria”. Esta cuestión obliga a mirar de frente a la rehabilitación, no como complemento tardío, sino como intervención con capacidad directa sobre los mecanismos que sostienen el dolor.
Referencias – Texto 1
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2-S15.
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895-926.
- Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Annu Rev Neurosci. 2018;41:459-486.
- Mayer EA. Gut/brain axis and the microbiota. J Clin Invest. 2015;125(3):926-938.
- Martin CR, Osadchiy V, Kalani A, Mayer EA. The brain-gut-microbiome axis. Cell Host Microbe. 2018;23(5):651-664.
- Sudo N, Chida Y, Aiba Y, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system. J Physiol. 2004;558:263-275.
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778-783.
- Donnelly CR, Andriessen AS, Chen G, et al. Central nervous system targets: glial mechanisms in chronic pain. Neurotherapeutics. 2020;17(3):846-860.